Xeroderma Pigmentosum and the XPA Gene
This page was produced as an assignment for Genetics 564, an undergraduate course at UW-Madison.
|
|
"...this [disease] has been a life and death battle with the sun since Cody was a baby. [Cody's mom] recalls the first time he was outside in the shade: "His entire skin was red, like somebody had just thrown grease on him. Every spot that had been exposed was freckles" "Cody is 7 years old, and the last time I brought him in to see the dermatologist, he said that Cody has the skin of a 70-year-old man."" --"The Real Story About Sun Protective Clothing" (at left)
|
Xeroderma pigmentosum (XP) is a rare genetic disorder that causes extreme sensitivity to ultraviolet rays (UV). In any individual, when excessive UV light hits the skin, it has the ability to produce genetic mutations in the DNA in skin cells. [1] Unaffected individuals possess a mechanism called nucleotide excision repair (NER), which recognizes these genetic mutations and removes them from the genome to prevent further damage. The sensitivity that XP individuals experience occurs as a result of a defective NER mechanism, and thus, an inability to repair the damage to DNA. Individuals with XP are also at a higher risk for damage by known carcinogens, such as cigarette smoke. As a result, these individuals are predisposed to many skin cancers, neurological damage, and other long-lasting effects.
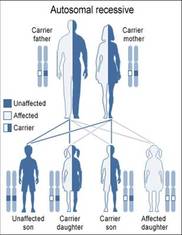
According to the Xeroderma Pigmentosum Society, prevalence
of this disorder is estimated at 1:1,000,000 in the United States. However, other populations, such as Japan, have a higher prevalence
(1:40,000). The disorder is especially prevalent in communities where consanguinity is common.
Currently, eight genes are known that cause XP: complementation group A (XPA) through complementation group G (XPG), as well as the variant type, XPV. [2] Xeroderma pigmentosum, complementation group
A (XPA), is known as the "classical form" of the disorder. At least 32
mutations are known in the XPA gene that cause XP. [3]
XP is an autosomal recessive disorder. This means that in order to show symptoms of the disease, an affected individual must have two defective copies of the XPA gene. Individuals who are heterozygous for the disorder will not show symptoms of the disorder, but may have an increased risk for skin cancer. [4] To produce an offspring with the disorder, both parents must have at least one copy of the gene; each offspring receives one copy of a gene from the mother and one from the father. In the figure, the couple has a 25% chance of having a child with XP.
XP is an autosomal recessive disorder. This means that in order to show symptoms of the disease, an affected individual must have two defective copies of the XPA gene. Individuals who are heterozygous for the disorder will not show symptoms of the disorder, but may have an increased risk for skin cancer. [4] To produce an offspring with the disorder, both parents must have at least one copy of the gene; each offspring receives one copy of a gene from the mother and one from the father. In the figure, the couple has a 25% chance of having a child with XP.
Signs and Symptoms
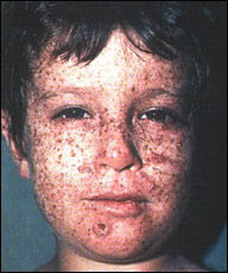
Symptoms of XP are evident during infancy or early childhood. The most distinctive features include blistering and redness of the skin with minimal sun exposure and lentigines (freckle-like spots, a distinctive quality of this disorder) on the face, shoulders, and any other areas that are exposed to sunlight. In some milder cases, individuals may tan normally, but lentigines are present in all cases by age two. This symptom rarely happens in children without the disorder. [2] The damage done by UV light causes premature aging, and the most affected areas are the skin, lips, tongue, and eyes. These areas are also at the highest risk for cancer. [5]
The eyes are heavily affected in XP patients. Photophobia, an abnormal visual intolerance to light, is common in patients with XP. The eyelids may be thin and flip inward or outward, or in severe cases, may be completely lost. [4]
Eyes also may become irritated and bloodshot if not protected, and the cornea may become cloudy. The risk of eye cancer is greatly increased in XP individuals, and the previously mentioned abnormalities may contribute to impaired vision or blindness. [2,5]
Neurological abnormalities are also associated with XP, and they are most commonly found in association with mutations in the XPA gene. [1] Neurological effects usually occur between ages three to eight years old. [7] These abnormalities can include developmental disabilities, mental retardation, difficulty walking, high hearing loss, difficulty swallowing and talking, and seizures. When these problems occur, they tend to worsen with time. [2, 5] Atypical cases have been reported in which XPA patients experience mild neurological complications, however, by adulthood, most XPA-afflicted individuals are bedridden due to their neurological problems. [7, 8] Avoidance of the sun and other forms of UV light does not prevent the progression of neurological deterioration. [9]
Without proper protection from the sun and other UV light, about half of children with XP will develop their first skin cancer by age 10. [2]
Children with XP have a greater than 10,000-fold increased risk of non-melanoma skin cancer at UV-exposed sites, and likely will develop skin cancers almost 60 years before the US general population. [4]
The eyes are heavily affected in XP patients. Photophobia, an abnormal visual intolerance to light, is common in patients with XP. The eyelids may be thin and flip inward or outward, or in severe cases, may be completely lost. [4]
Eyes also may become irritated and bloodshot if not protected, and the cornea may become cloudy. The risk of eye cancer is greatly increased in XP individuals, and the previously mentioned abnormalities may contribute to impaired vision or blindness. [2,5]
Neurological abnormalities are also associated with XP, and they are most commonly found in association with mutations in the XPA gene. [1] Neurological effects usually occur between ages three to eight years old. [7] These abnormalities can include developmental disabilities, mental retardation, difficulty walking, high hearing loss, difficulty swallowing and talking, and seizures. When these problems occur, they tend to worsen with time. [2, 5] Atypical cases have been reported in which XPA patients experience mild neurological complications, however, by adulthood, most XPA-afflicted individuals are bedridden due to their neurological problems. [7, 8] Avoidance of the sun and other forms of UV light does not prevent the progression of neurological deterioration. [9]
Without proper protection from the sun and other UV light, about half of children with XP will develop their first skin cancer by age 10. [2]
Children with XP have a greater than 10,000-fold increased risk of non-melanoma skin cancer at UV-exposed sites, and likely will develop skin cancers almost 60 years before the US general population. [4]
Treatment and Prevention

There is no cure for XP, and the damage that is done by UV rays is irreversible and cumulative. The best, and perhaps most obvious, strategy to manage this disorder is to avoid sunlight, other forms of UV rays, and known carcinogens. If an individual does need to be in the presence of UV light, the entire body must be fully protected. UV masks, as shown on the right, allow individuals to protect the face, eyes, and shoulders. Frequent skin examinations are recommended, as well as periodic routine eye and neurologic examinations and audiograms. [4]
References
1.Epstein, J., & Wang, S. Skin Cancer Foundation. http://www.skincancer.org/prevention/uva-and-uvb/understanding-uva-and-uvb. Retrieved February 3, 2014.
2. Genetics Home Reference. (2010). http://ghr.nlm.nih.gov/condition/xeroderma-pigmentosum. Retrieved January 27, 2014.
3. Amr, Khalda, Messaoud, Olfa, El Darouti, Mohamad, Abdelha, Sonia, El-Kamah, Ghada. (2014) Mutational specturm of Xeroderma pigmentosum group A in Egyptian patients. Gene, 533, 52-56. Available from http://www.ncbi.nlm.nih.gov/pubmed/24135642.
4. Kraemer KH, DiGiovanna JJ. Xeroderma Pigmentosum. 2003 Jun 20 [Updated 2013 Feb 14]. In: Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1397/ PubMed PMID: 20301571.
5. Xeroderma Pigmentosum Society. http://xps.org/wordpress/research-discovery/studentsresearch/. Retrieved February 3, 2014.
6. Swift, M., Chase, C. Cancer in families with xeroderma pigmentosum. J. Nat. Cancer Inst. 62: 1415-1421, 1979. Available from: http://www.ncbi.nlm.nih.gov/pubmed/286113 PubMed PMID: 286113.
7. Anttinen A, Koulu L, Nikoskelainen E, Portin R, Kurki T, Erkinjuntti M, Jaspers NG, Raams A, Green MH, Lehmann AR, Wing JF, Arlett CF, Marttila RJ. Neurological symptoms and natural course of xeroderma pigmentosum. Brain. 2008;131:1979–89. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18567921/ PubMed PMID: 18567921.
8. Robbins JH, Brumback RA, Mendiones M, Barrett SF, Carl JR, Cho S, Denckla MB, Ganges MB, Gerber LH, Guthrie RA. Neurological disease in xeroderma pigmentosum. Documentation of a late onset type of the juvenile onset form. Brain. 1991;114:1335–61. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2065254/ PubMed PMID: 2065254.
9. Ueda T, Kanda F, Aoyama N, Fujii M, Nishigori C, Toda T. Neuroimaging features of xeroderma pigmentosum group A. Brain Behav. 2012;1:1–5. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3343292/#b1 PubMed PMID: 22574268.
Note: All original sources for images on this website can be accessed by clicking on the image. Images without a link were created by Sarah Drewes.
1.Epstein, J., & Wang, S. Skin Cancer Foundation. http://www.skincancer.org/prevention/uva-and-uvb/understanding-uva-and-uvb. Retrieved February 3, 2014.
2. Genetics Home Reference. (2010). http://ghr.nlm.nih.gov/condition/xeroderma-pigmentosum. Retrieved January 27, 2014.
3. Amr, Khalda, Messaoud, Olfa, El Darouti, Mohamad, Abdelha, Sonia, El-Kamah, Ghada. (2014) Mutational specturm of Xeroderma pigmentosum group A in Egyptian patients. Gene, 533, 52-56. Available from http://www.ncbi.nlm.nih.gov/pubmed/24135642.
4. Kraemer KH, DiGiovanna JJ. Xeroderma Pigmentosum. 2003 Jun 20 [Updated 2013 Feb 14]. In: Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1397/ PubMed PMID: 20301571.
5. Xeroderma Pigmentosum Society. http://xps.org/wordpress/research-discovery/studentsresearch/. Retrieved February 3, 2014.
6. Swift, M., Chase, C. Cancer in families with xeroderma pigmentosum. J. Nat. Cancer Inst. 62: 1415-1421, 1979. Available from: http://www.ncbi.nlm.nih.gov/pubmed/286113 PubMed PMID: 286113.
7. Anttinen A, Koulu L, Nikoskelainen E, Portin R, Kurki T, Erkinjuntti M, Jaspers NG, Raams A, Green MH, Lehmann AR, Wing JF, Arlett CF, Marttila RJ. Neurological symptoms and natural course of xeroderma pigmentosum. Brain. 2008;131:1979–89. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18567921/ PubMed PMID: 18567921.
8. Robbins JH, Brumback RA, Mendiones M, Barrett SF, Carl JR, Cho S, Denckla MB, Ganges MB, Gerber LH, Guthrie RA. Neurological disease in xeroderma pigmentosum. Documentation of a late onset type of the juvenile onset form. Brain. 1991;114:1335–61. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2065254/ PubMed PMID: 2065254.
9. Ueda T, Kanda F, Aoyama N, Fujii M, Nishigori C, Toda T. Neuroimaging features of xeroderma pigmentosum group A. Brain Behav. 2012;1:1–5. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3343292/#b1 PubMed PMID: 22574268.
Note: All original sources for images on this website can be accessed by clicking on the image. Images without a link were created by Sarah Drewes.
Site Created By: Sarah Drewes
Contact: [email protected]
Last Modified: 05/18/14
University of Wisconsin-Madison
Contact: [email protected]
Last Modified: 05/18/14
University of Wisconsin-Madison
